Research concept
The general theme of our research is concerned with the issue of how small molecule ligands can interfere, act upon or modulate the properties and functions of proteins. Such processes involve the inhibition and/or modulation of enzymes, the activation or blocking of receptors or receptor complexes or the interference with signaling cascades via the perturbance of protein-ligand or protein-protein recognition. Fundamental to all these processes is the specific and selective recognition of the interacting biomolecules. Our research focuses on the following topics:- Analysis and classification
of structural data on protein-ligand complexes
- Development of experimental and computational
tools for the discovery and design of specific modulators of protein
function
- Experimental and computational
approaches towards a better understanding of binding affinity, selectivity and
interaction kinetics
- Design projects to modulate the function of proteins
relevant for different therapeutic areas such as infectious diseases or used
as engineered biocatalysts
We tackle the above-mentioned projects via a multi-disciplinary approach that involves the following techniques:
- Crystal structure analysis of proteins, ligands and protein-ligand
complexes
- Development of computational tools for data analysis, docking and
ligand design
- Thermodynamic and interaction kinetic characterization of protein-ligand
interactions by microcalorimetry and binding assays
- Expression, purification and mutagenesis of proteins, enzyme
kinetics
- Chemical synthesis of lead structures
DFG
EU
BMBF
ERC
CCDC
Synmikro
and different pharma companies which we would like to gratefully acknowledge
- Analysis and Classification of Structural Data on Protein-Ligand Complexes
- Relibase - an integrated database for protein-ligand complexes
Knowledge discovery from the exponentially growing body of structurally characterized protein-ligand complexes as a source of information in structure-based drug design is one of the major needs in contemporary drug research. Given the requirement for powerful data retrieval, integration and analysis tools, Relibase was developed as a database system particularly tailored to handle protein-ligand related problems (DOI, 133, DOI, DOI, 141). In many of our current projects, Relibase is used as routine tool for data analysis (133, 141) (collaboration: Cambridge Crystallographic Data Center (CCDC)).
- Waterbase - an analysis tool to study the influence of water on
ligand binding
Biological transformations are performed in aqueous solution, thus water is present as intimate partner in all ligand-binding processes. In two third of the known protein-ligand complexes water molecules mediate interactions between protein and ligand. Accordingly, a profound understanding and an adequate consideration of water molecules is a prerequisite for the prediction of ligand-binding modes, e.g. in docking. We have equipped Relibase with retrieval and analysis tools to study the properties of water molecules involved in ligand binding. Descriptors have been developed to predict the probability for the occurrence of water molecules in the protein-ligand binding interface (DOI).
Waterbase - an analysis tool to study the influence of water on ligand binding
- Cavbase - a database to compare binding pockets across
proteins
The function of proteins is almost invariably linked with the specific recognition of substrates and ligands in given binding pockets, thus proteins of related function should share comparable recognition properties exposed into these pockets. We have developed the new module Cavbase for Relibase that stores protein cavities in terms of simple surface-exposed physicochemical properties. These descriptors allow for fast retrieval of proteins with functional relationships independent of a particular sequence or fold homology (DOI, DOI). The approach also allows detecting unexpected cross-reactivity of ligands among unrelated proteins (DOI). Classification of binding pockets across protein family members allows elucidating selectivity determinants, e.g. in carbonic anhydrases, kinases, nuclear hormone receptors or proteases (DOI, DOI). Via spatial graph alignment, consensus binding epitopes are extracted and correlated to detect those physicochemical features that are conserved across individual members of protein families (DOI, DOI, 178, 215). This novel taxonomy has been applied to cluster the protein space of enzymes. The obtained clustering shows differences in the grouping based on sequence similarity but agrees with similarities in fold space or of the ligands hosted in the commonly structured binding pockets (PubMed). Clustering of cofactor binding pockets in cavity (Cavbase) and fold space (DALI) reveals virtually the same data structuring. Remarkable relationships can be found among the different spaces and show how conformations are conserved across the host proteins and which distinct local cavity and fold motifs recognize the different portions of the cofactors. In those cases, where different cofactors are found to be accommodated in a similar fashion to the same fold motifs, only a commonly shared substructure of the cofactors is used for the recognition process (DOI). Algorithms are developed for the automated analysis and decomposition of binding pockets in subpockets. Alternative approaches for the description of binding pockets by a set of surface patches to classify proteins (DOI) or new graph matching algorithms to cluster pockets have been developed (DOI) (funding; CCDC, DFG, collaboration: Prof. Hüllermeier Univ. Marburg, Prof. Ultsch, Univ. Marburg, CCDC, Sanofi).
Cavbase - a database to compare binding pockets across proteins
- Secbase - combining secondary structural information and folding
patterns with ligand binding data
The increasing body of structurally determined protein-ligand complexes indicates that particular secondary structural elements and folding motifs in proteins provoke preferred ligand-binding patterns. To search for such binding motifs possibly formed due to the recursive placement of physicochemical properties of secondary structural elements in special folding patterns of protein families, Relibase has been equipped with search facilities to combine information about helices, sheets and turns with ligand binding modes. An exhaustive classification of turn motifs has been performed and implemented into Relibase. It allows better coverage of turn motifs in fold prediction (DOI, DOI, DOI) (funding and collaboration: CCDC).
Secbase - combining secondary structural information and folding patterns with ligand binding data
- Analysis and interference of protein-protein interfaces
Most part of the presently known small molecule drugs are either enzyme inhibitors, allosteric effectors or receptor agonists or antagonists. They replace natural substrates or endogenous ligands mostly in deeply buried and stringent binding pockets. The presently available drug design tools are all methodologically focused on the competitive replacement of such ligands or substrate portions by appropriate lead structures that exploit a similar region of the deeply buried binding pocket. However, functional regulation of a biological system can also be achieved via inference with protein-protein interactions. In many processes, activation of protein function depends on co-activation through the assembly of several protein components, e.g. through the formation of a multi-domain complex. Many signal transduction cascades operate via the formation of protein-protein interfaces.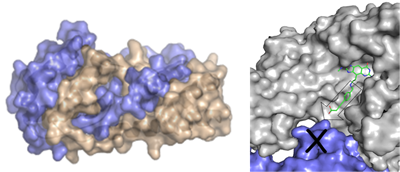
Analysis and interference of protein-protein interfaces
Accordingly, the interference with this recognition process using small molecule drugs would - in principle - allow for the development of an entirely new class of drugs. At present, we do not yet understand the nature and the driving force for the formation of protein-protein interactions, in particular with respect to their strength and dynamic stability. We have analyzed the structural patterns of exposed physicochemical properties exhibited by permanent and transient protein-protein interfaces (DOI). Also the reverse concept might interfere with information transduction: stabilization of protein-protein interactions. This can be achieved through accommodation of a small molecule ligand at the interface, thus enhancing the stability of a protein-protein contact (DOI) (collaboration: Prof. Hüllermeier, Univ. Marburg).
The tRNA modifying enzyme, tRNA-guanine transglycosylase (Tgt), a putative target for new selective antibiotics against Shigella bacteria, is only active as homodimer (see also 4b). By noncovalent mass spectrometry, we could confirm its dimeric oligomerisation state and a 2:1 binding stoichiometry of the complex formed between Tgt and its full-length substrate tRNA. To study whether dimer formation via a large protein-protein interface (>1600 Å2) is essential for Tgt to accomplish its catalytic function, point mutations were first investigated by a computational alanine scan. This approach indicates the residues most crucial for interface stabilization. Subsequently, the crucial residues have been mutated and enzyme kinetics reveal reduced catalytic activity of the mutated variants. The achieved destabilization toward monomer has been further evidenced by both noncovalent mass spectrometry and X-ray crystallography (DOI). On the basis of these results we currently try to develop interface inhibitors, which influence the dimer formation and antagonize the protein-protein interaction. This strategy displays a new approach to inhibit Tgt (collaborations: Prof. F. Diederich, ETH-Zürich, Switzerland, Dr. Sarah Sanglier-Cianferani and Prof. Alain Van Dorsselaer, Univ. Strasbourg, France, funding: DFG FO 806).
- Development of experimental and computational tools for the discovery and design of specific modulators of protein function
- Development of scoring functions for docking and affinity
prediction
Our group has been involved in the past in the development of computational tools for conformational analysis (PubMed, MIMUMBA), de novo design (DOI, LUDI), comparative molecular field analysis (PubMed, DOI, DOI, DOI, CoMSIA, AFMoC), comparative molecular superpositioning (PubMed, 73, DOI, DOI, FlexS) and ligand docking (DOI, FlexX). Key prerequisite in ligand docking (120, 122, 140) is, beside the generation of relevant binding modes, the correct estimate of binding affinity on the basis of the produced binding geometry (DOI, 122, PubMed, 140). Accordingly, we have developed the knowledge-based scoring function DrugScore (DOI) to rank different poses generated by docking methods. DrugScore employs statistically derived pair potentials using the distance-dependent occurence frequencies by which a particular ligand atom type is found in contact with a protein atom type. DrugScore can be tailored to a particular protein target using information about binding affinities of a small training set of ligands (DOI, AFMoC). As input, crystal data either from the PDB or the CSD are evaluated (DOI). Recently we could enhance DrugScore (DSX) by a novel atom type assignment and altered definitions of reference states (DOI). Furthermore, the DrugScore potentials can be used as objective function in ligand docking (DOI) and to optimize and refine the geometry of protein-ligand complexes (MiniMuDS, DOI). In addition, the regression-based scoring SFCscore has been developed that uses a broad range of different molecular descriptors as input. The analysis is performed on the basis of about 1000 protein-ligand complexes for which either affinity and structural data are available (DOI) (funding and collaboration: industry consortium, CCDC).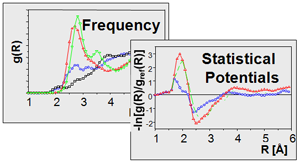
Development of scoring functions for docking and affinity prediction
- Hot-Spot analysis of binding sites
To discover favorable areas in a binding pocket, likely to accommodate a particular type of ligand functional group, we map the binding pocket in terms of "hot spots" of binding (DOI, 188). For this analysis, either Peter Goodford's force-field based method GRID can be applied, or knowledge-based approaches such as SuperStar and DrugScore (DOI, DOI, 188). The latter ones are both based on crystal data as input. They calculate preferred interaction sites by mapping either knowledge-based pair potentials or composite crystal-field environments onto active site-exposed residues (DOI). A regularly spaced grid is embedded into the binding site and for different ligand atom types interaction energies or contact preferences are calculated by systematically placing probe atoms at the various grid intersections. To allow for an intuitive graphical interpretation of the "hot spots", the obtained grid values are contoured according to a predefined level above the detected global minimum. Crucial in the analysis is an appropriate assignment of atom types to optimally represent the physicochemical properties of the "hot spot" under investigation. A sophisticated atom-type classification has been developed to extract knowledge-based potentials either from the PDB and CSD (DOI). From an experimental point of view, methods for cocrystallization and soaking of gases, solvent molecules and small molecular fragments are developed to perform an experimental active-site mapping.
Hot-Spot analysis of binding sites
- Strategies for Lead Discovery by Virtual Screening
Virtual screening (VS) is an alternative to high-throughput screening (HTS) in lead discovery. Hit selection is attempted in the computer by predicting binding properties of putative ligands (97, 162, DOI). The screened compounds do not necessarily exist and their testing does not consume valuable substance material. VS requires as key prerequisite knowledge about the three-dimensional structure of the target and the criteria responsible for ligand binding. It starts with a detailed analysis of the binding pocket of the target protein (Relibase). Using tools for hot-spot analysis, a protein-based pharmacophore is derived (PubMed, DOI, DOI, DOI, DOI, DOI).
Strategies for Lead Discovery by Virtual Screening
Subsequently, this information is used to define a list of essential functional groups to be present in putative candidate ligands. In the following, we apply a protocol of several consecutive hierarchical filters (PubMed, DOI, DOI) starting with a ligand preselection based on functional group requirements and fast pharmacophore matching. In subsequent steps, molecular similarity with known reference ligands is used to re-rank the hits from the pharmacophore matching. Finally, the best scored candidates are docked flexibly into the protein binding pocket.
We have applied virtual screening successfully to several protein targets (carbonic anhydrase II, thermolysin, trypsin, thrombin, tRNA guanine transglycosylase, peptide deformylase, aldose reductase, DOI, PubMed, DOI, DOI, DOI, DOI, DOI, DOI, DOI). The obtained computer hits gave rise to experimental testing of 10-15 compounds and nano- to micromolar binders were discovered. In several cases a crystal structure with the target protein could be determined (PubMed, DOI, DOI). Also in case of a homology-modelled GPCR receptor, our strategy retrieved an antagonist of submicromolar affinity. The considered model of the NK1 receptor was based on a newly developed approach which constructs proteins by homology including bound ligand molecules as restraints, thus resulting in more relevant geometries of protein binding sites (DOI, DOI, DOI, DOI). Initial homology models of the target protein are iteratively optimised by including information about bioactive ligands as spatial restraints (funding: DFG).
- Fragment-based Approaches to Lead Discovery
In recent times, methods are developed to discover small molecular fragments (MW < 250 Da) as first leads by a crystallographic screening. Once soaked or cocrystallized with a target protein, they provide first ideas about putative starting points for ligand development. As a kind of molecular probe, they explore the binding pocket and highlight the regions most favourable to accommodate small molecular fragments. At present, we extend our virtual screening protocols to compound libraries comprising candidates with a MW < 250 Da. Special scoring functions are required together with methodologically enhanced docking tools. Furthermore, we improve experimental soaking techniques to achieve a higher success rate in getting small molecular fragments into protein crystals. For this purpose, the group develops special exposure techniques using a free mounting device for crystals which allows manipulating crystals freely accessible in a humidity gas stream (collaboration: Proteros, BioSolveIt, Merck, Boehringer Ingelheim, funding: BMBF).
Fragment-based Approaches to Lead Discovery
- Experimental and computational approaches towards a better understanding of binding affinity, selectivity and interaction kinetics
- Microcalorimetry to better understand binding affinity in
protein-ligand complexes
Binding affinity is an essential entity to predict the potency of a ligand in structure-based drug design. We recently suggested to start ligand optimization with hits exhibiting largest enthalpic efficacy (DOI). Isothermal titration calorimetry (ITC) gives access to the thermodynamic signature of the overall ligand binding event, however, which additional effects are overlaid and what are the net criteria to pick the ligand with the best enthalpic binding properties, means the one with the largest enthalpic efficiency?
While docking programs have meanwhile achieved a level of reliability that makes them a viable tool for database screening of possible leads on the computer, the ranking of putative hits according to their expected affinity remains the most crucial step in this procedure (67, PubMed, DOI, 105, PubMed, DOI). Accordingly, there is an acute need for a better understanding what "binding affinity" really means for the recognition of a drug at its receptor and how this binding relates to thermodynamics and binding kinetics. Using isothermal titration calorimetry, protein crystallography, molecular dynamics simulations and biophysical methods to determine interaction kinetics, we study the binding of series of low molecular-weight ligands towards model proteins such as trypsin, thrombin, thermolysin, carbonic anydrase, HIV protease, endothiapepsin, SAP II, protein kinase A, tRNA guanine transglycosylase and aldose reductase (DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI). In congeneric series of ligands, surprising changes of protonation states can occur (DOI, DOI). They originate from induced pKa shifts experienced by the ligand and protein functional groups upon complex formation (induced dielectric fit). They depend on the local environment and oxidation state of bound cofactors and involve additional heat effects that must be corrected before any conclusion on the binding enthalpy (ΔH) and entropy (ΔS) can be drawn. To complement the experimental evidence, we apply computer simulations to predict changes of protonation states. These calculations involve free energy calculations and we developed a uniform charge model either for the ligands and protein residues (DOI, DOI, DOI, DOI).
Microcalorimetry to better understand binding affinity in protein-ligand complexes
After correction, trends in both contributions can be interpreted in structural terms with respect to the hydrogen-bond inventory or residual ligand and protein motions or the change in the local water structure. Even across congeneric series, the factorization in enthalpy and entropy can change significantly, usually in a way that both contributions mutually compensate each other, leaving the free energy of binding ΔG virtually unchanged. Explanation for these compensating effects are changes in the local water structure (pick-up or release of water molecules), differences in the residual mobility of protein residues or portions of the ligands, an unbalanced solvation/desolvation inventory and shifts in the local charge distribution giving rise to modified charge assisted hydrogen bonds (DOI, DOI). Pronounced cooperativity effects are in operation that demonstrate the inadequacy of simple additivity models to describe functional group contributions to binding affinity (DOI, DOI, DOI, DOI, DOI, DOI) (collaboration: Prof. A. Podjarny, CNRS, Illkirch, France; Prof. D. Hangauer, SUNY Buffalo, USA, funding: DFG).
- Site specific mutagenesis among members of a protein family to study
selectivity determinants
In order to investigate the issue of selectivity and specificity in protein-ligand interactions from an experimental point of view, we study the step-wise reconstruction of binding pockets in related proteins being members of a particular protein family. As a first example, the binding site of human factor Xa has been introduced into the structurally related rat and bovine trypsins by site-directed mutagenesis (DOI, DOI). Major re-organisation of the binding site takes place to yield a geometry virtually identical to that of factor Xa. However, with respect to binding affinity, still a significant difference is observed between factor Xa and the trypsin variants. To achieve closer relationship in the binding properties, also residues located in the second coordination sphere around the binding pocket have to be exchanged (DOI, DOI, DOI, DOI, DOI). As a second example we selected aldose and aldehyde reductase. Both enzymes possess similar binding pockets, however, aldehyde reductase exhibits an additional loop, comprising 11 residues, that is responsible for differences in substrate specificity. In this part, aldose reductase opens a hydrophobic specificity pocket and shows pronounced adaptations upon ligand binding (DOI). Binding of two chemically closely related Aldose Reductase inhibitors has been studied against a series of single-site mutants of the wild-type protein. Overall, the binding mode of the inhibitors is conserved; but tiny structural changes are responded by partly strong modulation of the thermodynamic profiles and pronounced enthalpy/entropy compensations. This provides insights how single-site mutations can alter selectivity of closely related ligands against a target protein (DOI) (collaboration: Prof. M. Schlitzer, Univ. Marburg, funding: DFG).
Site specific mutagenesis among members of a protein family to study selectivity determinants
- 3D-QSAR analysis and clustering of binding cavities to elucidate
affinity and selectivity determinants
Computational approaches to analyze selectivity start with the comparison of binding affinities across a set of structurally diverse ligands with respect to several members of a protein family. By means of 3D QSAR methods, such as comparative molecular field analyses, molecular properties of ligands can be extracted that determine protein binding. If this evaluation is focused on affinity differences, the mutual comparison of the derived QSAR models allows one to elucidate the most important selectivity determinants (DOI, DOI, DOI, DOI). Complementary to this evaluation of ligand data, binding pockets across different members of a protein family can be analyzed. Generalized probes are used that explore the properties exposed by the protein towards the binding-site cavity (cf. GRID, SuperStar, DrugScore or CavBase descriptors and surface patches). They provide a set of functionally similar descriptors and can be correlated via chemometric and clustering analyses (DOI, DOI, DOI, DOI). In case of Cavbase, the binding pocket-exposed physicochemical properties are computed and stored in a similarity matrix. This matrix serves as input for a hierarchical cluster analysis. It unravels structural similarities among binding pockets, not apparent through sequence information. Proteins distant in sequence space can be clustered together due to close relationship in cavity space. Close similarity in the latter space indicates possible cross-reactivity of putative ligands and suggests where to expect conflicting selectivity profiles (DOI, DOI, PubMed) (funding: DFG, CCDC, Novartis).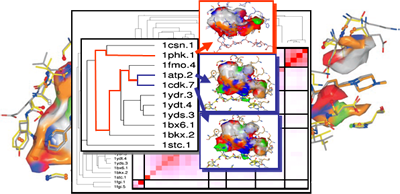
3D-QSAR analysis and clustering of binding cavities to elucidate affinity and selectivity determinants
- Design Projects on Protein Targets relevant for Different Therapeutic Areas such as Infectious Diseases or as Engineered Biocatalysts
- De novo crystal structure determination of proteins, involved in
infectious and other diseases, structure-based ligand design
Several crystal structures of novel proteins as putative targets for drug therapy have been determined, e.g., 1-deoxy-D-xylulose-5-phosphate reductoisomerase, a crucial enzyme in the non-mevalonate isoprenoid biosynthesis (DOI), the Plasmodium falciparum glutamate dehydrogenase, a putative target for novel antimalaria drugs (DOI), lipoamide dehydrogenase from Trypanosoma cruzi, a target for Chagas disease therapy, the tRNA-modifying enzyme S-adenosylmethionine:tRNA ribosyl transferase/Isomerase, a possible target for Shigella Dysentry (DOI).
Structure-based ligand design is performed on transglutamminase TG2, a putative target for Coeliac disease, an autoimmune disorder of the small intestine that occurs in genetically predisposed people (collaboration: Zedira, Darmstadt, funding: BMBF). A virtual screening campaign against bacterial peptide deformylase (PDF) discovered several hits that are currently evaluated as putative leads to inhibit this class of proteins to combat infectious diseases such as malaria along this concept (collaboration: Prof. M. Schlitzer, Univ. Marburg).
- Inhibitor design of t-RNA transglycosylase: A putative target for
Shigellosis therapy
Shigellae are the causative agents of dysentery (Shigellosis) and effect more than one million deaths each year. Usually Shigellosis is treated by antibiotics but since more and more multidrug-resistant strains are reported, there is urgent need for the development of new antibiotics. Characterisation of chromosomal mutants of S. flexneri has resulted in the identification of a gene, that contributes significantly to pathogenicity and codes for tRNA-guanine transglycosylase (TGT). This enzyme, involved in biosynthesis of the highly modified nucleoside queuine inserted in the anticodon loop of certain tRNAs, has been recognized as essential in the regulation of bacterial virulence. Accordingly, it has been selected as a target for structure-based inhibitor design. Based on the crystal structure of TGT with a bound substrate, virtual screening has discovered a series of small molecule hits subsequently found to bind in the micromolar range. Several of these initial hits served as first lead for an iterative optimization to compounds with improved binding affinity. Cycles of design, synthesis, and crystal structure analysis revealed pronounced induced fit adaptations of the enzyme with respect to bound ligands. The importance of a conserved water network, not to be perturbed by ligand binding, could be evidenced. Significant shift in binding affinity could be detected by moving from a normal to charge-assisted hydrogen binding. Interestingly, a set of nanomolar inhibitors shows pronounced residual mobility of one side chain of the inhibitors. This entropically favourable contribution to binding seems beneficial for ligand binding (DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI, 221, DOI). Kinetic and mutational studies have elucidated the substrate selectivity profile of TGT originating from different species. Structural modifications of the bacterial enzyme toward the human protein could be performed to better understand the differences in substrate specificity between the species (DOI, DOI, DOI) (collaboration: Prof. F. Diederich, ETH-Zürich, Switzerland, Prof. G. Garcia, Univ. Michigan, USA, funding: DFG, NIH).
Inhibitor design of t-RNA transglycosylase: A putative target for Shigellosis therapy
- Design of inhibitors against family members of serine and aspartyl proteases
Mining the human genome for possible drug targets has revealed pronounced clustering of proteins into several prominent families. Likely, these families share family-wide commonalities in terms of enzymatic mechanisms and molecular recognition properties. This, in consequence, suggests that priviledged ligand scaffolds can be designed to address entire families. To achieve specificity and selectivity, proper decoration of the underlying privileged scaffolds is required. Through combinatorial chemistry, a given molecular scaffold can be easily modified with respect to the decorating side-chains. An appropriate selection of these side-chains is achieved by the detailed analysis of the recognition properties of the various subsites that compose the binding pocket accommodating the scaffold. Taking the families of serine and aspartyl proteases as case studies, synthetic and computational tools are developed how to optimize a given privileged ligand scaffold towards different family members. The side-chain selection step is supported by hot-spot analyses and similarity searches with Cavbase. Once a binding pocket similar to the query pocket is detected, the accommodated ligand might suggest an alternative molecular portion suitable as decoration of the privileged scaffold under investigation (DOI, DOI, DOI).
Design of inhibitors against family members of serine and aspartyl proteases therapy
HIV- protease, plasmepsin II and IV, pepsin and SAP II have been selected as target proteins for structure-based design. Through several cycles of iterative design, first leads could be optimized with respect to affinity, selectivity and resistance tolerance profile. A novel scaffold could be detected for HIV protease that shows, compared to all currently available market products, a deviating binding mode. It exhibits a different resistance profile and shows higher affinity towards one of the major resistance mutants compared to the wildtype (DOI, DOI, DOI, DOI, DOI, DOI, DOI, DOI) (collaboration: Prof. W. Diederich, Univ. Marburg, funding: DFG).
- Structural studies on Shigella-specific pathogenicity factors as a basis for structure-based drug design
The bacterial invasion of Shigella, the causative agent of bacillary dysentery, depends on its ability to invade colonic epithelial cells via self induced macropinocytosis. Invasion of the host cell involves a number of massive cytoskeletal rearrangements within the colonic epithelial cell which are mainly triggered by the so-called invasines. These effectors secreted by the bacterium cause the recruitment of cytoskeletal proteins as well as actin polymerisation at the site of contact with the bacterium. The cytoskeletal rearrangements during Shigella invasion and intracellular spread are regarded as a model system for common processes during cell movement and adhesion. As there is only few biochemical and structural information available about the proteins causing these dynamical changes, we started to isolate and crystallize the involved bacterial effectors in order to determine their 3D structures. With a view to the understanding of their mechanisms of action at a molecular level, we aim to establish the crystal structures of these proteins not only in their apo-forms but also in complex with known protein interaction partners. Not least, these pathogenicity factors will represent highly interesting targets for the structure-based design of drugs against bacillary dysentery (collaboration: Prof. K. Reuter, Univ. Marburg, funding: DFG).
- Design of substrate specificity of biocatalysts, hybrid enzymes, catalysts for click chemistry
The binding pocket of a protein provides an anisotropic environment well suited to perform selective and highly stereospecific chemical reactions. For example, lipases catalyze the hydrolysis, transesterification or amidation of a broad range of esters and amides with distinct stereopreference. In consequence of their stability and large scale availability, they have found widespread applications in the enantioselective synthesis of precursors to pharmaceuticals and in the kinetic resolution of racemic mixtures. We have studied the kinetic resolution of preacylated Candida antarctica lipase by enzyme kinetics and crystallography along with computer simulations. We could evidence that the fast-reacting enantiomer is enthalpically favored through a virtually perfect active-site complementarity, whereas the slow-reacting substrate compensates some of the discriminating free enthalpy advantage by a beneficial entropic contribution. This is due to a higher residual mobility and thus a smaller loss in entropy upon binding. It resides less frequently in an orientation productive for the enzyme reaction (DOI) (collaboration and funding: BASF).
By tailored design we modify protein structures in a way to catalyze novel chemical reactions. As an example, the Huisgen reaction which produces triazole and tetrazole heterocycles via 1,3-dipolar addition has been selected. This reaction is either CuI- or ZnII-catalyzed. In consequence, the zinc ion in thermolysin has been exchanged by copper and we try to incorporate a second metal ion centre into carbonic anhydrase via site-directed mutagenesis (DOI). As reaction components, small libraries of substituted alkynes or nitrils, and azides are used. The azide component could be tethered to the enzyme surface via a disulfide bridge, while the alkyne component was reversibly coordinated via a sulfon amide anchor to the zinc ion in the literal catalytic center of the enzyme. The incipient orientation of the reactants in the binding site and of the formed triazole product were characterized by crystallography and the reaction progression could be monitored by HPLC-MS analysis (DOI) (collaboration: Prof. U. Koert, Univ. Marburg, funding: DFG-FO 594).
Design of substrate specificity of biocatalysts, hybrid enzymes, catalysts for click chemistry therapy
Abbildungen:
1b: Judith Günther, Doktorarbeit, Universität Marburg, 2003.
1c: Daniel Kuhn, Doktorarbeit, Universität Marburg, 2004.
1d: Oliver Koch, Doktorarbeit, Universität Marburg, 2008.
2a: Holger Gohlke, Doktorarbeit, Universität Marburg, 2001.
2c: https://upload.wikimedia.org/wikipedia/commons/9/91/MTT_Plate.jpg, abgerufen am 24.01.2017/12:00 Uhr.
4b: Kristallstruktur der tRNA Guanintransglycosylase TGT mit gebundener tRNA (PDB-Code: 1Q2S), Dr. Mathias Zentgraf.
top of the page