What is drug design?
An introduction for non-specialists
Probably everyone has already taken medication. Perhaps some of you have asked yourself what actually happens to my body when the tablet dissolves and the substances it contains are released. Why does the aspirin find the headache and can free me from this pain?
After ingestion, a tablet begins to dissolve in the stomach or intestine and releases the drug it contains, usually a low-molecular organic compound. The compound then overcomes the wall of the stomach and intestines and enter the bloodstream. From there, the transport through the body begins, which initially leads via the liver. The aim of this transport is to find the place where a drug is to take its effect. These places are usually receptors or biochemical catalysts (enzymes) that perform important functions in our body. For example, receptors are located on the surface of cells, and by binding a small molecule a signal is transmitted to the cell, so that processes are set in operation inside the cell. The body itself produces such signaling substances, such as adrenaline. Drugs can now compete with these messenger substances for the receptors and thus influence the tasks of a cell by either triggering or blocking processes in a targeted manner. Enzymes, biochemical catalysts in our body are responsible for the conversion of substances in our body. With their help, the building blocks that we need for all the processes of life are synthesized and chemically transformed; examples are the metabolism of our food in our intestine or processes that ensure that a bleeding wound is closed again.
The ingredient of aspirin, acetylsalicylic acid, also attacks such an enzyme and influences its function. It plays an important role in the biosynthesis of the body's own compounds (so-called inflammation mediators), which are increasingly formed in inflamed tissue and, among other things, sensitize the pain receptors. Aspirin is able to reduce the formation of these compounds.
In order for a drug substance to influence the function of an enzyme, it must bind specifically and selectively to this catalyst. The site where the body's own substances are normally converted must be blocked. This task can be compared with the function of a key in a lock (Fig. 1 left). Only if a key has the right shape and form, it fits into the lock and can perform its function. Drugs that act as enzyme inhibitors do have the correct fit of the key, but they override the function of the lock. This can be compared to breaking the key in the lock.
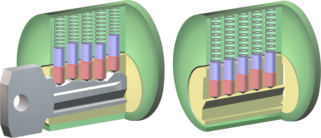
Fig. 1
The drug researcher now tries to find optimal keys for a given lock. This search for the right keys for the given lock is the subject of our research. What is necessary? First, the three-dimensional geometry of the lock must be known. The spatial structure of enzymes and receptors can be elucidated with X-ray crystallography. To do this, these very large biomolecules must be crystallized. In a crystal, a large number of these molecules come together and arrange themselves regularly next to each other. Crystals are in turn capable of diffracting X-rays. From the generated diffraction images, it is then possible to calculate the structural makeup of the molecules that have generated the diffraction image of the crystal.
We have set up a computer-controlled device at our institute or we measure at a synchrotron source, which can be used to carry out X-ray diffraction experiments on crystals, and use it to determine the structures of our locks. Of course, this does not yet provide the shape and properties of suitable keys to fit to the lock. However, if such a key is available, it is straight-forward to determine its binding geometry, means how it is arranged in the lock, again by crystal structure analysis of the lock, but now together with the bound key (Fig. 2).
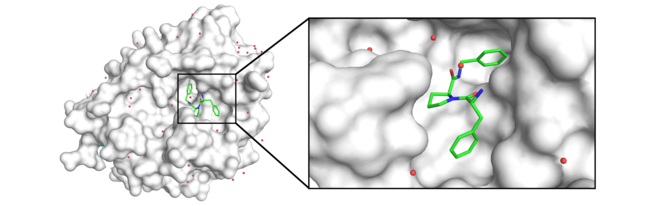
Fig. 2
The search for new keys is the main focus of our research group. On the one hand, one can start from the shape of the binding pocket of the enzyme or receptor and use this template to find new keys (Fig. 2 right). On the other hand, one can take large collections of keys, i.e. databases of molecules, and try one key after the other to see if it fits into the given lock. This complex analysis can only be done with a computer; therefore, we develop docking programs that support us in this task. These search procedures require an understanding of the criteria that are responsible for a key actually being suitable for a given lock. As nice as the comparison of key and lock is, it simplifies the conditions given by interacting molecules. Molecules are conformationally flexible, they can build up different kinds of interactions. All these aspects must be correctly understood for the search to be successful. Unfortunately, we are only just beginning to compile the criteria and to understand them gradually. Therefore, we also conduct experiments that give us information about the criteria that ultimately determine why a drug binds to a certain protein. At the same time, we have to create new molecules that have emerged as ideas from our computer searches. We carry out this work primarily with colleagues who have the synthesis of new compounds as the focus of their work. If we want to understand why a certain key fits, we try to put the cart before the horse in our key search. Not only can we modify the structure of our keys, we can also do this with our locks. The binding pockets in enzymes and receptors are composed of amino acids. These amino acids can be exchanged and it is possible to see how this replacement affects the binding of a drug. At the same time, we learn more about the function of a biomolecule. We carry out the exchanges (mutations) in a targeted manner. Also Nature often makes use of these changes (evolution). For example, functionally identical enzymes and receptors from humans, various animals or bacteria differ by such exchanges of amino acids. Furthermore, resistance to certain drugs is created precisely by such exchanges. For our key search it is very important to understand the criteria on which the selectivity differences between similar keys are based or how resistance to potent drugs is generated. For example, a drug is intended to affect the enzymes of a bacterium or virus, but the human enzymes must remain unaffected. Here no general key is helpful, here a selective special key must be available.
It is not possible to elucidate the structure of every lock. Nevertheless, even in these cases keys are needed as potential drugs. On the one hand, we can try to find new keys by comparing already known keys, which may not be optimal in some respects. Also the structures of different keys, which all fit into an unknown but common lock, contain a lot of information about this lock (Fig. 3). This information must be extracted and used.

Fig. 3
On the other hand one can also try to build a model of the unknown protein binding site. Models can only be designed if you have certain ideas about how the lock should look like, i.e., you must know already related locks. This can only be understood if you can screen the existing data. After all, the crystal structures of about 170,000 proteins have already been elucidated today. For this reason, our research group is developing a database in which it is possible to search for structures of enzymes and receptors, but also to ask questions to small molecules bound to these proteins. By analyzing these data we want to find out whether there are similarities in the shape of the binding pockets that take up small molecules, e.g. drugs. We hope that we will then be able to use these findings for the search and targeted design of new drugs.
Illustrations:
Fig1: CC BY-SA 3.0, retrieved on 24.01.2017, 12:00
https://de.wikipedia.org/wiki/Schlie%C3%9Fzylinder#/media/File:Pin_tumbler_no_key.svg
https://de.wikipedia.org/wiki/Schlie%C3%9Fzylinder#/media/File:Pin_tumbler_with_key.svg
Fig2: Crystal structure thrombin (PDB: 2ZFF)
Baum, B., Mohamed, M., Zayed, M., Gerlach, C., Heine, A., Hangauer, D., & Klebe, G. (2009). More than a Simple Lipophilic Contact: A Detailed Thermodynamic Analysis of Nonbasic Residues in the S1 Pocket of Thrombin. Journal of Molecular Biology, 390, 56-69. http://doi.
Seitenanfang